Frontiers in Genetics
Introduzione
I processi cellulari sono guidati da più molecole interagenti il cui livello di attività deve essere regolato dinamicamente (Kitano, 2002). Di conseguenza, i geni appartenenti alla stessa via di segnalazione e metabolica o che condividono funzioni simili tenderanno ad essere co-espressi tra le condizioni (Wang et al., 2016). L’analisi del modulo genico di co-espressione crea reti che comprendono insiemi di geni (cioè moduli) la cui espressione è altamente correlata. Tale analisi è stata applicata per rivelare moduli funzionali relativi a infettivi (Janova et al., 2015), infiammatorio (Beins et al., 2016), e neurologico (Voineagu et al., 2011) malattie, così come diversi tipi di cancro (Sharma et al., 2017).
Weighted gene co-expression network analysis (WGCNA) è un metodo ampiamente utilizzato per identificare i moduli genici co-espressi (Zhang e Horvath, 2005). Per eseguire WGCNA, tuttavia, gli utenti devono avere familiarità con gli ambienti di programmazione e selezionare manualmente i parametri. Queste caratteristiche impediscono ai ricercatori con una conoscenza insufficiente di R di identificare i moduli genici dai set di dati del trascrittoma.
Basato sul nostro pacchetto Bioconductor R denominato CEMiTool (Russo et al., 2018), abbiamo sviluppato un’applicazione basata sul Web user-friendly che consente agli scienziati senza background in bioinformatica di eseguire un’analisi completa della rete di co-espressione.
Materiali e metodi
L’interfaccia web di webCEMiTool è stata sviluppata per consentire agli utenti di generare rapidamente analisi complete senza la necessità di installare alcun programma specifico o browser internet. L’unico requisito per eseguire l’analisi modulare è un set di dati contenente i livelli di espressione di tutti i geni in campioni in diverse condizioni biologiche (qui definite come “classi”). Non esiste un numero di campioni definito, ma il nostro studio precedente suggerisce un minimo di campioni 15 per set di dati (Russo et al., 2018). Sebbene sia stato progettato principalmente per i dati del trascrittoma (cioè RNA-seq o microarray), può anche essere potenzialmente utilizzato per identificare moduli di proteine, citochine e persino metaboliti. webCEMiTool selezionerà quindi automaticamente i geni di input e identificherà i moduli di co-espressione. Ogni modulo contiene un insieme di geni la cui espressione segue un modello simile.
Abbiamo implementato, all’interno di webCEMiTool, una funzionalità che valuta l’attività dei moduli genici su ogni classe di campioni. Per questo, gli utenti devono solo fornire un file di testo delimitato da tabulazioni di annotazione di esempio che informa la classe di ciascun campione. Un “diagramma di profilo” che mostra il livello mediano dei singoli geni all’interno del modulo viene quindi visualizzato nella sezione “Risultati” dello strumento (Figura 1A).
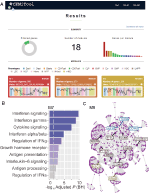
Figura 1. Panoramica di webCEMiTool. (A) Sintesi dei risultati di webCEMiTool – Il grafico a ciambella rappresenta la proporzione di geni selezionati dal filtro non supervisionato. La prima pagina mostra anche il numero di moduli ottenuti, così come un grafico a barre raffigurante il numero di geni in ogni modulo. I diagrammi del profilo del modulo illustrano l’attività di espressione mediana dei geni dai moduli attraverso ogni campione. I colori rappresentano le diverse classi di esempio. (B) Analisi di sovrarappresentazione – Questo descrive il valore p regolato −log10 (Benjamini-Hochberg) dei percorsi arricchiti in un modulo (percorsi definiti dall’utente immesso .file gmt). (C) Rete genica di un modulo – I geni più collegati (hub) sono etichettati e colorati in base al fatto che fossero originariamente presenti nel modulo (blu), o inseriti da un file di interazione immesso dall’utente (rosso), o entrambi (verde).
Per consentire l’analisi funzionale, gli utenti possono anche verificare se i moduli genici sono associati a specifiche vie di segnalazione o metaboliche (Figura 1B). Questi percorsi possono essere facilmente estratti da database, come KEGG, Reactome e MySigDB. Infine, gli utenti possono integrare i risultati con i dati interactome (cioè, interazioni proteina-proteina, fattori di trascrizione e loro geni trascritti, o anche miRNA e loro geni bersaglio). Questa funzione consente agli utenti di identificare i regolatori critici dei moduli (Figura 1C), fornendo informazioni preziose per la convalida sperimentale o potenziali obiettivi per i farmaci. Ulteriori dettagli su come ottenere i file opzionali sono disponibili nella pagina “Tutorial” del sito web.2
Per dimostrare che il nostro metodo è robusto, abbiamo eseguito un’analisi modulare su larga scala senza precedenti con oltre 1.000 set di dati RNA-seq e microarray disponibili pubblicamente e nuovi dati RNA-seq di pazienti infetti da Leishmania utilizzando la versione del pacchetto CEMiTool R (Russo et al., 2018). Sebbene webCEMiTool e il pacchetto abbiano caratteristiche di visualizzazione distinte e siano basati su piattaforme diverse, la funzionalità di co-espressione principale è essenzialmente la stessa. Lo strumento online che stiamo descrivendo qui è costruito per consentire un facile accesso alle analisi modulari dei geni per i ricercatori non programmanti,mentre la versione della libreria R è orientata verso gli utenti con una maggiore conoscenza del linguaggio di programmazione R. Inoltre, la dashboard dei risultati è composta da grafici interattivi che facilitano l’interpretazione. Inoltre, sfruttando il crescente ecosistema di servizi web bioinformatici, il nostro strumento stabilisce un’interfaccia con la piattaforma Enrichr (Chen et al., 2013), consentendo un’esperienza più ricca per i nostri utenti.
Risultati
Abbiamo dimostrato che webCEMiTool può essere applicato per analizzare i dati di espressione a livello di singola cella. I dati disponibili pubblicamente di viscRNA-Seq (virus-compreso RNA a cella singola-Seq) sono stati ottenuti dal database NCBI GEO (accession number GSE110496) e utilizzati come input per l’analisi. I dati si riferiscono al trascrittoma di singole cellule di epatoma umano (Huh7), che sono state infettate dal virus dengue (DENV) o dal virus Zika (ZIKV), utilizzando la molteplicità di infezione (MOI) 0, 1 o 10 (Zanini et al., 2018). Le cellule raccolte su quattro diversi punti temporali (4, 12, 24 e 48 h dopo l’infezione) sono state quindi ordinate per l’analisi trascrittomica a singola cellula con un protocollo Smart-seq2 adattato (Zanini et al., 2018). Il set di dati DENV comprende 933 cellule infette (MOI = 1 o 10) e 303 controlli (MOI = 0), mentre il set di dati ZIKV è composto da 488 cellule infette (MOI = 1) e 403 controlli. Prima di inviare l’analisi alla piattaforma webCEMiTool, entrambi i set di dati sono stati trasformati log10 e i geni che non erano espressi in più dell ‘ 80% dei campioni sono stati rimossi. I set di dati sono stati quindi divisi per virus e per punto di tempo e utilizzati come input (campo”Expression file”) in webCEMiTool. Oltre ai dati di espressione genica, abbiamo anche fornito a webCEMiTool i fenotipi del campione (es., carichi virali) e insiemi del gene del Reactome.
Le nostre analisi webCEMiTool hanno generato una media di sei moduli per punto temporale nell’infezione da DENV e più di otto moduli per punto temporale nell’infezione da ZIKV. Abbiamo selezionato un modulo per punto temporale come rappresentante dei nostri risultati (Figura 2A). È chiaro che a 24 e 48 h post-infezione, l’attività di espressione dei moduli rappresentativi aumenta in base alla carica virale (Figura 2A). Successivamente abbiamo eseguito l’analisi di arricchimento del percorso dei moduli rappresentativi a 24 h post-infezione utilizzando il link webCEMiTool per Enrichr (Figura 2B). Questi risultati non solo confermano ciò che è stato descritto nella pubblicazione originale (Zanini et al., 2018) ma anche fornire nuove intuizioni sulla fisiopatologia delle infezioni da virus dengue e Zika.
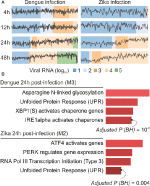
Figura 2. webCEMiTool applicato ai dati RNA-seq a cella singola. (A) Profilo grafico dei moduli genici co-espressi. Abbiamo selezionato un modulo rappresentativo per ogni infezione da virus post-dengue (a sinistra) o infezione da virus post-Zika (a destra). La linea nera rappresenta l’attività di espressione mediana dei geni dai moduli attraverso ogni campione. I colori rappresentano la diversa quantità di RNA virale all’interno della cellula. (B) Analisi di sovrarappresentazione di moduli selezionati a 24 ore dopo l’infezione da virus. I grafici a barre sono stati adattati dal webtool Enrichr collegato a webCEMiTool. Le barre sono proporzionali al valore p regolato-log10 (Benjamini-Hochberg) dei percorsi arricchiti in un modulo.
Discussione
Sebbene siano state sviluppate poche applicazioni simili basate sul Web per eseguire analisi geniche di co-espressione (Tzfadia et al., 2016; Desai et al., 2017), questi strumenti non forniscono risultati comparabili a webCEMiTool. Una di queste applicazioni è GeNET (Desai et al., 2017). Questo webtool è stato progettato per facilitare le analisi di co-espressione genica e fornisce analisi di arricchimento e reti gene-to-gene. Tuttavia, esegue solo queste analisi per tre organismi (R. capsulatus, M. tuberculosis e O. sativa). Un altro esempio è CoExpNetViz (Tzfadia et al., 2016), un webtool progettato per la visualizzazione e la costruzione di reti geniche. Simile a GeNET, CoExpNetViz è un po ‘ limitato rispetto agli organismi come si afferma di essere progettato principalmente per trascrittomi vegetali. Il webCEMiTool mira a fornire analisi di co-espressione per qualsiasi organismo. Inoltre, sebbene CoExpNetViz sia presentato come un’applicazione basata sul Web, i suoi risultati vengono restituiti agli utenti come una cartella compressa contenente un README.file txt con le istruzioni su come visualizzare i loro risultati sul Cytoscape app. Gli utenti devono quindi inserire manualmente in Cytoscape i diversi file di output forniti dallo strumento. Questi passaggi aggiuntivi possono anche rendere il processo soggetto a errori e forse scoraggiante per gli utenti che non hanno familiarità con Cytoscape. Il webCEMiTool offre risultati visualizzati dal browser molto più convenienti.
Abbiamo anche dimostrato che webCEMitool è in grado di analizzare i dati RNA-seq a cella singola in modo più rapido ed efficiente. I nostri risultati hanno restituito informazioni rilevanti sui processi biologici coinvolti con l’infezione da virus dengue e Zika. Tutte queste analisi sono state eseguite in modo automatizzato e pratico, senza la necessità per l’utente di avere una profonda comprensione sull’elaborazione interna dell’analisi dei dati di co-espressione genica.
Contributi dell’autore
LC, PR, BG-C e MA-P hanno eseguito le analisi. Il sito web è stato sviluppato da LC, GS-H e VM-C. HN ha concepito lo strumento e supervisionato il lavoro. Tutti gli autori aiutano nella scrittura del documento.
Dichiarazione sul conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.