Frontiers in Genetics
Introduksjon
Cellulære prosesser drives av flere samvirkende molekyler hvis aktivitetsnivå må reguleres dynamisk (Kitano, 2002). Som et resultat vil gener som tilhører samme signalering og metabolsk vei eller deling av lignende funksjoner, ha en tendens til å bli uttrykt på tvers av forhold (Wang et al., 2016). Samekspresjonsgenmodulanalyse skaper nettverk som består av sett med gener (dvs. moduler) hvis uttrykk er svært korrelert. Slik analyse ble anvendt for å avdekke funksjonelle moduler relatert til smittsomme (Janova et al., 2015), inflammatorisk (Bein et al., 2016), og nevrologisk (Voineagu et al., 2011) sykdommer, samt flere typer kreft (Sharma et al., 2017).
Vektet gene co-expression network analysis (WGCNA) er en mye brukt metode for å identifisere co-uttrykt genmoduler(Zhang Og Horvath, 2005). For å kunne kjøre WGCNA må brukerne imidlertid være kjent med programmeringsmiljøer, samt å manuelt velge parametere. Disse funksjonene hindrer forskere med utilstrekkelig kunnskap Om R for å identifisere genmoduler fra transkriptomdatasett.
Basert på Vår Bioconductor R pakke kalt Cemitool (Russo et al., 2018), utviklet vi en brukervennlig nettbasert applikasjon som gjør det mulig for forskere uten bakgrunn i bioinformatikk å utføre omfattende samuttrykksnettverksanalyse.
Materialer Og Metoder
webgrensesnittet til webCEMiTool ble utviklet for å tillate brukere å raskt generere omfattende analyser uten behov for å installere noe bestemt program eller nettleser. Det eneste kravet for å kjøre den modulære analysen er et datasett som inneholder uttrykksnivåene for alle gener i prøver under forskjellige biologiske forhold (her definert som “klasser”). Det er ingen definert utvalg antall prøver, men vår tidligere studie antyder et minimum av 15 prøver per datasett (Russo et al ., 2018). SELV om det primært ble designet for transkriptomdata (dvs.RNA-seq eller mikroarrays), kan det også potensielt brukes til å identifisere moduler av proteiner, cytokiner og til og med metabolitter. webCEMiTool vil da automatisk velge inngangsgener og identifisere samekspresjonsmodulene. Hver modul inneholder et sett med gener hvis uttrykk følger et lignende mønster.
vi implementerte, innenfor webCEMiTool, en funksjon som vurderer aktiviteten til genmoduler på hver klasse av prøver. For dette må brukerne bare gi en eksempelannonsering tabulatordelt tekstfil som informerer klassen av hver prøve. Et “profilplott” som viser mediannivået for individuelle gener i modulen, vises deretter i” Resultater ” – delen av verktøyet (Figur 1a).
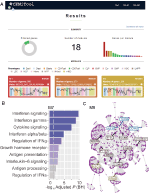
Figur 1. webCEMiTool oversikt. (A) webCEMiTool resultater sammendrag-donut diagrammet representerer andelen av utvalgte gener av uten tilsyn filter. Forsiden viser også antall moduler som er oppnådd, samt et stolpediagram som viser antall gener i hver modul. Modulprofilplott illustrerer median ekspresjonsaktiviteten til gener fra modulene over hver prøve. Fargene representerer de forskjellige utvalgsklassene. (B) overrepresentasjonsanalyse-dette viser-log10-justert p-verdi −Benjamini-Hochberg) av de berikede banene i en modul (pathways definert av bruker-inputted .gmt-fil). (C) Gennettverk av en modul – de øverste mest tilkoblede gener (huber) er merket og farget basert på om de opprinnelig var tilstede i modulen (blå), eller satt inn fra en brukerinnstilt interaksjonsfil (rød), eller begge deler (grønn).
for å muliggjøre funksjonsanalyse kan brukerne også sjekke om genmodulene er knyttet til spesifikke signalerings-eller metabolske veier (Figur 1b). Disse banene kan enkelt hentes fra databaser, slik SOM KEGG, Reactome, Og MySigDB. Endelig kan brukerne integrere resultatene med interactome data (dvs.protein-protein interaksjoner, transkripsjonsfaktorer og deres transkriberte gener, eller til og med mirna og deres målgener). Denne funksjonen gjør det mulig for brukere å identifisere kritiske regulatorer av moduler (Figur 1c), og gir verdifull innsikt for eksperimentell validering eller potensielle mål for narkotika. Ytterligere detaljer om hvordan du får de valgfrie filene kan bli funnet i” Tutorial ” side av nettstedet.2
for å demonstrere at vår metode er robust, utførte vi en enestående storskala modulær analyse med over 1000 offentlig tilgjengelige RNA-seq-og microarray-datasett og nye rna-seq-data fra pasienter infisert Med Leishmania ved Hjelp Av cemitool r-pakkeversjonen (Russo et al., 2018). Selv om webCEMiTool og pakken har forskjellige visualiseringsfunksjoner og er basert på forskjellige plattformer, er kjerneko-uttrykksfunksjonaliteten i det vesentlige den samme. Det elektroniske verktøyet vi beskriver her er bygget for å muliggjøre enkel tilgang til genmodulære analyser for ikke-programmerende forskere, mens r library-versjonen er rettet mot brukere med større kunnskap Om r-programmeringsspråket. I tillegg er resultatene dashbordet består av interaktive diagrammer som letter tolkning. Videre utnytter det stigende økosystemet av bioinformatikk webtjenester, vårt verktøy etablerer et grensesnitt Med Enrichr-plattformen (Chen Et al., 2013), som muliggjør en rikere opplevelse for våre brukere.
Resultater
vi viste at webCEMiTool kan brukes til å analysere uttrykksdata på enkeltcellenivå. Offentlig tilgjengelige viscRNA-Seq-data (virus-inkludert enkeltcelle RNA-Seq) ble hentet fra NCBI GEO database (tiltredelsesnummer GSE110496) og brukt som input for analysen. Dataene refererer til transkriptomet av individuelle humane hepatom (Huh7) celler, som var infisert med enten DENGUE virus (DENV) eller Zika virus (ZIKV), ved bruk av multiplikasjon av infeksjon (MOI) 0, 1 eller 10 (Zanini et al., 2018). Celler samlet på fire forskjellige tidspunkter (4, 12, 24 og 48 h etter infeksjon) ble deretter sortert for enkeltcelle transkriptomisk analyse med en tilpasset Smart-seq2-protokoll (Zanini et al., 2018). DENV – datasettet består av 933 infiserte celler (MOI = 1 eller 10) og 303 kontroller (MOI = 0), MENS ZIKV-datasettet består av 488 infiserte celler (MOI = 1) og 403 kontroller. Før analysen ble sendt til webCEMiTool-plattformen, ble begge datasettene log10 transformert og gener som ikke ble uttrykt i mer enn 80% av prøvene ble fjernet. Datasettene ble deretter delt av virus og etter tidspunkt og brukt som input (“Expression file” felt) til webCEMiTool. I tillegg til genuttrykk data, vi også gitt til webCEMiTool prøven fenotyper (dvs., viral belastning) Og Reaktom gensett.
våre webCEMiTool-analyser genererte i gjennomsnitt seks moduler per tidspunkt I DENV-infeksjon og mer enn åtte moduler per tidspunkt I ZIKV-infeksjon. Vi har valgt en modul per tidspunkt som representant for våre funn (Figur 2a). Det er klart at ved 24 og 48 h etter infeksjon øker ekspresjonsaktiviteten til representative moduler i henhold til virusbelastningen (Figur 2a). Vi utførte deretter pathway enrichment analysis av representative modulene ved 24 timer etter infeksjon ved hjelp av webCEMiTool-lenken For Enrichr (Figur 2b). Disse funnene bekrefter ikke bare det som ble beskrevet i den opprinnelige publikasjonen (Zanini et al., 2018) men gir også ny innsikt om fysiopatologien til dengue-og Zika-virusinfeksjoner.
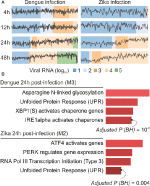
Figur 2. webCEMiTool brukes på enkeltcelle RNA-seq data. (A) Profilplott av co-uttrykte genmoduler. Vi valgte en representativ modul for hvert tidspunkt post-dengue virus infeksjon (venstre) eller post-Zika virus infeksjon (høyre). Den svarte linjen representerer median ekspresjonsaktiviteten til gener fra modulene over hver prøve. Fargene representerer den forskjellige mengden virus RNA i cellen. (B) overrepresentasjonsanalyse av utvalgte moduler ved 24 timer etter virusinfeksjon. Søylediagrammer ble tilpasset Fra Enrichr webtool knyttet til webCEMiTool. Stolpene er proporsjonale med −log10 justert p-verdi (Benjamini-Hochberg)av de berikede banene i en modul.
Diskusjon
selv om noen lignende web – baserte applikasjoner ble utviklet for å utføre co-uttrykk genanalyse (Tzfadia et al ., 2016; Jørgen et al., 2017), gir disse verktøyene ikke sammenlignbare resultater med webCEMiTool. En slik søknad er GeNET (Desai Et al., 2017). Denne webtool er designet for å lette genet co-uttrykk analyser og gir berikelse analyse og gene-til-gene nettverk. Imidlertid utfører den bare disse analysene for tre organismer(r. capsulatus, m. tuberculosis og O. sativa). Et annet eksempel Er CoExpNetViz (Tzfadia et al., 2016), en webtool designet for visualisering og bygging av gennettverk. I likhet Med GeNET, CoExpNetViz er noe begrenset med hensyn til organismer som det sies å være primært utformet for plante transkriptomes. WebCEMiTool har som mål å gi samuttrykksanalyser for enhver organisme. Videre, Selv Om CoExpNetViz presenteres som en web – basert applikasjon, blir resultatene returnert til brukere som en komprimert mappe som inneholder EN README.txt fil med instruksjoner om hvordan å visualisere sine resultater På Cytoscape app. Brukerne har da å manuelt sette Inn Cytoscape flere forskjellige utdatafiler levert av verktøyet. Disse ekstra trinnene kan også gjøre prosessen utsatt for feil og muligens skremmende for brukere som ikke er kjent Med Cytoscape. Den webCEMiTool tilbyr mye mer praktisk nettleser viste resultater.
vi viste også at webCEMitool er i stand til å analysere enkeltcelle RNA-seq data raskere og effektivt. Våre resultater returnerte relevant informasjon om de biologiske prosessene som er involvert med dengue og Zika virusinfeksjon. All denne analysen ble utført på en automatisert og praktisk måte, uten behov for at brukeren skal ha dyp forståelse for den interne behandlingen av gendataekspresjonsanalyse.
Forfatterbidrag
LC, PR, BG-C og MA-P utførte analysene. LC, GS-H og VM-C utviklet webtool. HN unnfanget verktøyet og overvåket arbeidet. Alle forfattere hjelper til med å skrive papiret.
Interessekonflikt
forfatterne erklærer at forskningen ble utført i fravær av kommersielle eller økonomiske forhold som kan tolkes som en potensiell interessekonflikt.