Frontiers in Genetics
wprowadzenie
procesy komórkowe są napędzane przez wiele oddziałujących ze sobą cząsteczek, których poziom aktywności musi być dynamicznie regulowany (Kitano, 2002). W rezultacie geny należące do tego samego szlaku sygnałowego i metabolicznego lub dzielące podobne funkcje będą miały tendencję do współwystępowania w różnych warunkach (Wang et al., 2016). Co-expression gene module analysis tworzy sieci zawierające zestawy genów (to znaczy, moduły) których ekspresja jest wysoce skorelowana. Taka analiza została zastosowana do ujawnienia funkcjonalnych modułów związanych z infekcją (Janova et al., 2015), zapalne (Beins et al., 2016) i neurologiczne (Voineagu et al., 2011) choroby, a także kilka rodzajów raka (Sharma et al., 2017).
Weighted gene co-expression Network analysis (WGCNA) jest powszechnie stosowaną metodą identyfikacji współwyrażonych modułów genowych (Zhang and Horvath, 2005). Aby jednak uruchomić WGCNA, użytkownicy muszą być zaznajomieni ze środowiskami programistycznymi, a także ręcznie wybierać parametry. Cechy te uniemożliwiają badaczom z niewystarczającą znajomością R identyfikację modułów genowych z zestawów danych transkryptomowych.
w oparciu o nasz pakiet Bioconductor R o nazwie CEMiTool (Russo et al., 2018), opracowaliśmy przyjazną dla użytkownika aplikację internetową, która umożliwia naukowcom bez doświadczenia w bioinformatyce wykonywanie kompleksowej analizy sieci koekspresji.
materiały i metody
interfejs webowy webCEMiTool został opracowany tak, aby umożliwić użytkownikom szybkie generowanie kompleksowych analiz bez konieczności instalowania konkretnego programu lub przeglądarki internetowej. Jedynym wymaganiem do przeprowadzenia analizy modułowej jest zbiór danych zawierający poziomy ekspresji wszystkich genów w próbkach w różnych warunkach biologicznych (zdefiniowanych w niniejszym dokumencie jako “klasy”). Nie ma zdefiniowanej liczby próbek zakresu, ale nasze poprzednie badania sugerują co najmniej 15 próbek na zestaw danych (Russo et al., 2018). RNA-seq lub mikromacierzy), może być również potencjalnie używany do identyfikacji modułów białek, cytokin, a nawet metabolitów. webCEMiTool automatycznie wybierze geny wejściowe i zidentyfikuje Moduły koekspresji. Każdy moduł zawiera zestaw genów, których ekspresja przebiega według podobnego schematu.
zaimplementowaliśmy w webCEMiTool funkcję, która ocenia aktywność modułów genowych w każdej klasie próbek. W tym celu użytkownicy muszą tylko dostarczyć przykładowy plik tekstowy rozdzielony tabulatorami adnotacji, który informuje klasę każdej próbki. “Wykres profilu” pokazujący medianę poziomu poszczególnych genów w module jest następnie wyświetlany w sekcji “wyniki” narzędzia (rysunek 1a).
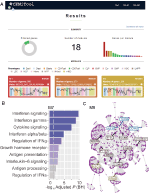
Rysunek 1. przegląd webCEMiTool. A) podsumowanie wyników webCEMiTool-wykres przedstawia proporcję wybranych genów przez filtr nienadzorowany. Strona główna wyświetla również liczbę uzyskanych modułów, a także wykres słupkowy przedstawiający liczbę genów w każdym module. Wykresy profilu modułu ilustrują medianę aktywności ekspresji genów z modułów w każdej próbce. Kolory reprezentują różne przykładowe klasy. B) analiza Nadreprezentacyjna-przedstawia skorygowaną wartość p-log10 (Benjamini-Hochberg) wzbogaconych ścieżek w module (ścieżki zdefiniowane przez użytkownika-inputted .gmt file). (C) Sieć genów modułu – najlepsze najbardziej połączone geny (Huby) są oznaczone i zabarwione na podstawie tego, czy były pierwotnie obecne w module (niebieski), czy wstawione z pliku interakcji wprowadzonego przez użytkownika (czerwony), czy oba (zielony).
aby umożliwić analizę funkcjonalną, użytkownicy mogą również sprawdzić, czy moduły genów są związane ze specyficznymi szlakami sygnałowymi lub metabolicznymi (rysunek 1B). Ścieżki te można łatwo wyodrębnić z baz danych, takich jak KEGG, Reactome i MySigDB. Wreszcie, użytkownicy mogą zintegrować wyniki z interactome danych (to znaczy, interakcje białko-białko, czynniki transkrypcyjne i ich transkrypcji genów, lub nawet Mirna i ich genów docelowych). Funkcja ta umożliwia użytkownikom identyfikację krytycznych regulatorów modułów (rysunek 1C), dostarczając cennych informacji na potrzeby eksperymentalnej walidacji lub potencjalnych celów dla leków. Dodatkowe informacje na temat uzyskiwania opcjonalnych plików można znaleźć na stronie “Tutorial”na stronie internetowej.2
aby wykazać, że nasza metoda jest solidna, przeprowadziliśmy bezprecedensową analizę modułową na dużą skalę z ponad 1000 publicznie dostępnych zestawów danych RNA-seq i mikromacierzy oraz nowe dane RNA-seq pacjentów zakażonych Leishmania przy użyciu wersji pakietu CEMiTool R (Russo et al., 2018). Chociaż webCEMiTool i pakiet mają różne funkcje wizualizacji i są oparte na różnych platformach, podstawowa funkcja koekspresji jest zasadniczo taka sama. Narzędzie online, które tutaj opisujemy, zostało stworzone w celu umożliwienia łatwego dostępu do modułowych analiz genów dla naukowców spoza programowania, natomiast wersja biblioteki R jest skierowana do użytkowników z większą znajomością języka programowania R. Dodatkowo pulpit wyników składa się z interaktywnych wykresów, które ułatwiają interpretację. Ponadto, korzystając z rosnącego ekosystemu bioinformatycznych usług internetowych, nasze narzędzie ustanawia interfejs z platformą Enrichr (Chen et al., 2013), umożliwiając bogatsze wrażenia dla naszych użytkowników.
wyniki
pokazaliśmy, że webCEMiTool może być stosowany do analizy danych ekspresji na poziomie pojedynczej komórki. Publicznie dostępne dane viscRNA-Seq (wirus-w tym RNA jednokomórkowego-Seq) uzyskano z bazy danych NCBI GEO (numer akcesyjny GSE110496) i wykorzystano jako dane wejściowe do analizy. Dane odnoszą się do transkryptomu poszczególnych ludzkich komórek hepatoma (Huh7), które zostały zainfekowane wirusem dengi (DENV) lub wirusem Zika (ZIKV), wykorzystując wielość infekcji (MOI) 0, 1 lub 10 (Zanini i wsp., 2018). Komórki zebrane w czterech różnych punktach czasowych (4, 12, 24 i 48 h po zakażeniu) zostały następnie posortowane do analizy transkryptomicznej pojedynczych komórek za pomocą zaadaptowanego protokołu Smart-seq2 (Zanini et al., 2018). Zestaw danych DENV składa się z 933 zainfekowanych komórek (MOI = 1 lub 10) i 303 kontrolek (MOI = 0), podczas gdy zestaw danych ZIKV składa się z 488 zainfekowanych komórek (MOI = 1) i 403 kontrolek. Przed przesłaniem analizy do platformy webCEMiTool, oba zestawy danych zostały przekształcone log10 i usunięto geny, które nie zostały wyrażone w więcej niż 80% próbek. Zbiory danych zostały następnie podzielone przez wirusa i punkt czasowy i użyte jako wejście (pole” Expression file”) do webCEMiTool. Oprócz danych ekspresji genów, dostarczyliśmy również webCEMiTool przykładowe fenotypy (tj., wiremia) i Reaktomowych zestawów genów.
nasze analizy webCEMiTool wygenerowały średnio sześć modułów na punkt czasowy w zakażeniu DENV i ponad osiem modułów na punkt czasowy w zakażeniu ZIKV. Wybraliśmy jeden moduł na punkt czasowy jako przedstawiciel naszych ustaleń (rysunek 2A). Oczywiste jest, że po 24 i 48 h po zakażeniu aktywność ekspresyjna reprezentatywnych modułów wzrasta w zależności od miana wirusa (Fig.2A). Następnie wykonaliśmy analizę wzbogacania szlaku dla reprezentatywnych modułów w 24 h po zakażeniu przy użyciu łącza webCEMiTool dla Enrichr (ryc. 2b). Te ustalenia nie tylko potwierdzają to, co zostało opisane w oryginalnej publikacji (Zanini et al., 2018), ale także dostarczają nowych informacji na temat fizjopatologii zakażeń wirusem dengi i Zika.
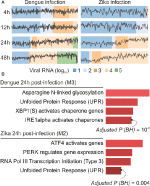
Rysunek 2. webCEMiTool zastosowany do jednokomórkowych danych RNA-seq. A) Wykres profilu modułów genów współwyrażonych. Wybraliśmy jeden reprezentatywny moduł dla każdego punktu czasowego po zakażeniu wirusem dengi (po lewej) lub po zakażeniu wirusem Zika (po prawej). Czarna linia reprezentuje medianę aktywności ekspresji genów z modułów w każdej próbce. Kolory reprezentują różną ilość RNA wirusa w komórce. B) Analiza Nadreprezentacji wybranych modułów w ciągu 24 h po zakażeniu wirusem. Wykresy słupkowe zostały zaadaptowane z WebTool enrichr połączonego z webCEMiTool. Pręty są proporcjonalne do SKORYGOWANEJ-log10 wartości p (Benjamini-Hochberg) ścieżek wzbogaconych w module.
dyskusja
chociaż opracowano kilka podobnych aplikacji internetowych do wykonywania analizy genów koekspresji (Tzfadia et al., 2016; Desai et al., 2017), narzędzia te nie zapewniają porównywalnych wyników z webCEMiTool. Jednym z takich zastosowań jest GeNET (Desai et al., 2017). Ten webtool został zaprojektowany w celu ułatwienia analizy koekspresji genów i zapewnia analizę wzbogacania i sieci genów do genów. Jednak przeprowadza te analizy tylko dla trzech organizmów (R. capsulatus, M. tuberculosis i O. sativa). Innym przykładem jest CoExpNetViz (Tzfadia et al., 2016), webtool przeznaczony do wizualizacji i budowy sieci genowych. Podobnie jak GeNET, CoExpNetViz jest nieco ograniczony w odniesieniu do organizmów, ponieważ twierdzi się, że jest przeznaczony głównie do transkryptomów roślinnych. WebCEMiTool ma na celu dostarczenie analiz koekspresji dla każdego organizmu. Co więcej, chociaż CoExpNetViz jest prezentowany jako aplikacja internetowa, jego wyniki są zwracane użytkownikom w postaci skompresowanego folderu zawierającego README.plik txt z instrukcjami, jak wizualizować swoje wyniki w aplikacji Cytoscape. Użytkownicy muszą następnie ręcznie wstawić do Cytoscape kilka różnych plików wyjściowych dostarczonych przez narzędzie. Te dodatkowe kroki mogą również sprawić, że proces będzie podatny na błędy i może zniechęcać użytkowników nieznających Cytoscape. WebCEMiTool oferuje znacznie wygodniejsze wyniki wyświetlane przez przeglądarkę.
pokazaliśmy również, że webCEMitool jest w stanie szybciej i wydajniej analizować dane RNA-seq jednokomórkowego. Nasze wyniki zwróciły istotne informacje na temat procesów biologicznych związanych z zakażeniem dengą i wirusem Zika. Wszystkie te analizy zostały przeprowadzone w sposób zautomatyzowany i praktyczny, bez potrzeby, aby użytkownik miał głębokie zrozumienie wewnętrznego przetwarzania analizy danych koekspresji genów.
LC, PR, BG-C i MA-P wykonały analizy. LC, GS-H I VM-C opracowały webtool. HN wymyślił narzędzie i nadzorował pracę. Wszyscy autorzy pomagają w pisaniu pracy.
Oświadczenie o konflikcie interesów
autorzy oświadczają, że badanie zostało przeprowadzone przy braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.