Aplicación de ensayos de complementación de fragmentos de proteínas en biología celular
- Introducción
- Principio
- Limitaciones
- Controles estándar para un Estudio de PCA
- Aplicación de PCA en el Diseño de proteínas: Selección de Bibliotecas vs. Bibliotecas para Proteínas que Interactúan de manera Óptima
- Aplicación de PCA al Cribado de la Biblioteca de ADNc en Células de mamíferos
- Usando PCA como Regla Molecular: Estudios de receptores
- Mapeo de redes bioquímicas
- Conclusión
- Agradecimientos
Introducción
Las vías bioquímicas son realmente sistemas de ensamblaje y desmontaje dinámico de complejos de proteínas, y por lo tanto, gran parte de la investigación biológica moderna se ocupa de cómo, cuándo y dónde las proteínas interactúan con otras proteínas involucradas en procesos bioquímicos. La demanda de enfoques simples para estudiar las interacciones proteína-proteína, particularmente a gran escala, ha aumentado recientemente con el progreso de los proyectos de genoma, ya que la asociación de productos genéticos desconocidos con productos genéticos conocidos proporciona una forma crucial de establecer la función de un gen. Con este desafío en mente, nuestro laboratorio desarrolló ensayos de complementación de fragmentos de proteínas (PCA). En esta estrategia, dos proteínas de interés (proteínas A y B) se fusionan con fragmentos complementarios de una proteína reportera (una enzima, una proteína fluorescente, etc.).). Si las proteínas A y B interactúan, los fragmentos del reportero se unen, se pliegan en la estructura nativa del reportero y reconstituyen su actividad (Figura 1). Las proteínas reporteras de PCA han sido elegidas como aquellas que producen una variedad de actividades detectables, incluyendo señales fluorescentes, luminiscentes y colorimétricas, así como ensayos de selección de supervivencia simples (1-14). Hemos demostrado que la estrategia de PCA tiene las siguientes capacidades: (i) permite la detección de interacciones proteína-proteína in vivo e in vitro en cualquier tipo de célula; (ii) permite la detección de interacciones proteína-proteína en compartimentos subcelulares u orgánulos apropiados; (iii) permite la detección de interacciones inducidas específicamente en respuesta a señales de desarrollo, nutricionales, ambientales o inducidas por hormonas; (iv) permite el monitoreo de los aspectos cinéticos y de equilibrio del ensamblaje de proteínas en las células; y (v) permite la detección de nuevas interacciones proteína-proteína en cualquier tipo de célula (2,3,6,9) (15-19).
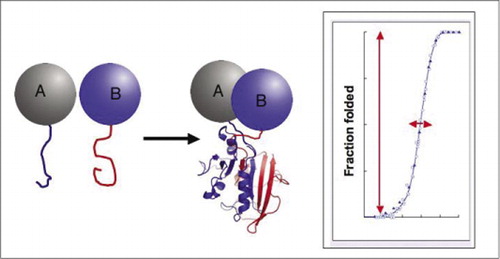
Si las dos proteínas interactúan, los fragmentos de la proteína reportera se unen, se pliegan en la estructura nativa de la proteína reportera y su actividad se reconstituye (izquierda). Estos ensayos de complementación de fragmentos de proteínas (PCA) tienen características físicas que los hacen particularmente útiles como reporteros de complejos dinámicos de proteínas. A la derecha hay una curva de plegado de proteínas donde el eje x es un parámetro variable (p. ej., concentración de un fragmento en relación con otro). La alta cooperatividad de este proceso (aumento extremadamente agudo en la fracción de especies plegadas en un rango muy estrecho) significa que los ensayos tienen un rango dinámico enorme, lo que hace que la detección de un complejo sea un fenómeno virtual de todo o nada. Esto contrasta con métodos como la transferencia de energía de resonancia de fluorescencia (FRET), que tiene un rango dinámico muy bajo y requiere una optimización cuidadosa de una serie de parámetros. Por el contrario, medir la formación de complejos proteicos mediante PCA no es más difícil que medir la actividad de la enzima reportera intacta.
Principio
Demostramos el principio del PCA comenzando con la enzima dihidrofolato reductasa (DHFR) como reportero (1). Era obvio que si el plegamiento de la enzima a partir de sus fragmentos (como se detecta por reconstitución de actividad) dependía absolutamente de la unión de las proteínas que interactúan, entonces el sistema descrito es, de hecho, un detector de las interacciones. Nosotros y otros hemos demostrado desde entonces que este principio se puede generalizar a una serie de enzimas, incluidas las luciferasas de Gaussia y Renilla, la β-lactamasa TEM, así como la proteína fluorescente verde (GFP) y sus variantes (1-14). Una característica crucial de los fragmentos de PCA es que están diseñados para no plegarse espontáneamente sin ser acercados por la interacción de las proteínas a las que se fusionan (1,20). Si se produjera un plegado espontáneo, el PCA simplemente no funcionaría. El plegado espontáneo conduciría a una señal positiva falsa, una situación que confundiría irremediablemente la interpretación de las pantallas de biblioteca in vivo (que se espera que sea una aplicación importante). En contraste con el PCA, hay sistemas de ensayo basados en β-galactosidasa e inteínas divididas que se asemejan al PCA, pero que son conceptual y prácticamente diferentes (21,22). En ambos casos, las bien conocidas subunidades de las enzimas que ocurren naturalmente y se asocian espontáneamente se fusionan con proteínas que interactúan. El problema central aquí es que las subunidades, incluso si se asocian débilmente, siempre son capaces de hacerlo hasta cierto punto, lo que significa que hay un trasfondo constante de ensamblaje espontáneo.
Limitaciones
La estrategia de PCA es general, en el sentido de que no se limita a un solo reportero enzimático, y ha sido ideada en varias formas diferentes, cada una de las cuales es más adecuada para abordar una pregunta específica. Por ejemplo, los PCA simples de selección de supervivencia, como los basados en DHFR, son más útiles para la selección de bibliotecas, mientras que los PCA de lectura de luminiscencia o fluorescencia son los mejores para estudios de la dinámica espacial y temporal de complejos proteicos. Debido a que las proteínas de fusión se pueden expresar en células que son modelos relevantes para estudiar una ruta bioquímica específica, es probable que estén en su estado biológico nativo, incluidas las modificaciones postraduccionales correctas (obviamente, los fragmentos de PCA en sí no deben interferir con la orientación o modificación de las proteínas, y esto debe ser probado).
Entre los PCA más simples y por lo tanto más populares se encuentran los basados en proteínas fluorescentes (como GFP y variantes), porque la señal es proporcionada por el fluoróforo intrínseco (7-9)(14,15,17,23). Sin embargo, las proteínas fluorescentes deben expresarse a altos niveles para asegurar que la señal esté por encima de la fluorescencia celular de fondo, y se ha demostrado que las proteínas fluorescentes PCA son irreversibles, lo que puede ser útil (atrapando y visualizando complejos raros), pero también podría conducir a una mala interpretación del recambio o la localización de proteínas que interactúan (8,23,24). Por otro lado, se ha demostrado que los PCA basados en DHFR y β-lactamasa como reporteros, basados en evidencia indirecta, son reversibles después de la interrupción de las interacciones, mientras que un PCA basado en Gaussia luciferasa ha demostrado directamente ser reversible (2,3,6). Por lo tanto, la reversibilidad del PCA permite la detección de aspectos cinéticos y de equilibrio del ensamblaje y desmontaje de complejos proteicos en células vivas (Figura 2).
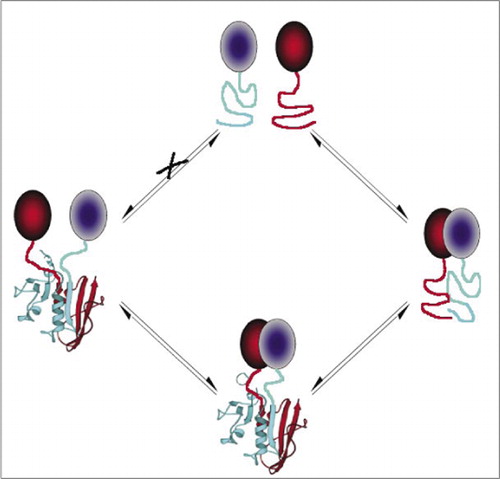
Esto evita la asociación espontánea de los fragmentos (vía X) que puede conducir a una señal falsa. Igualmente, se seleccionan fragmentos para los que el despliegue espontáneo de fragmentos debe ocurrir cuando se interrumpe el complejo proteico (lado izquierdo).
Controles estándar para un Estudio de PCA
La estrategia de PCA requiere que los fragmentos de la proteína reportera se ensamblen y se plieguen después de que las proteínas de interés hayan formado un complejo. El ensamblaje y plegado correcto del reportero depende de la recuperación tanto de la geometría estructural intrínseca a las proteínas del reportero como del complejo formado por las proteínas que interactúan. Esta es una de las principales distinciones de los ensayos de PCA en comparación con los ensayos de transferencia de energía de resonancia de fluorescencia (FRET) o transferencia de energía de resonancia de bioluminiscencia (BRET) o los ensayos de dos híbridos de levadura, y esta característica nos permitió realizar un estudio basado en la estructura del receptor de eritropoyetina (19). Por lo general, insertamos un enlazador de polipéptidos flexibles de 10 aminoácidos que consiste en (Gly.Gly.Gly.Gly.Ser) 2 entre la proteína de interés y el fragmento reportero de PCA (para ambas fusiones). Este enlazador se eligió porque es el más flexible posible, y hemos observado empíricamente que los enlazadores de esta longitud son lo suficientemente largos como para permitir que los fragmentos se encuentren y se plieguen, independientemente del tamaño de las proteínas que interactúan a las que se fusionan los fragmentos (16).
Para garantizar que no se produzcan respuestas inespecíficas, se debe realizar un conjunto de controles. Estos controles podrían incluir los siguientes, aunque el primero es el más importante: (i) Proteínas que no interactúan. No se debe observar una respuesta al PCA si se utilizan proteínas que no interactúan como socios del PCA; la sobreexpresión de una proteína que no interactúa por sí sola tampoco debería competir por la interacción conocida. ii) Mutaciones en la interfaz de proteínas asociadas. Una mutación puntual o de deleción de una pareja que se sabe que interrumpe una interacción también debe prevenir una respuesta a la PCA. iii) Competencia. Una respuesta de PCA debe ser disminuida por la sobreexpresión simultánea de una u otra de las proteínas que interactúan y que no están fusionadas con un fragmento de PCA complementario. iv) Intercambio de fragmentos. Una interacción observada entre dos proteínas debe ocurrir incluso si las proteínas se intercambian con los fragmentos de reportero respectivos.
Aplicación de PCA en el Diseño de proteínas: Selección de Bibliotecas vs. Bibliotecas para Proteínas que Interactúan de manera Óptima
Entre las primeras aplicaciones de un PCA se encontraba un problema de diseño de proteínas. El ensayo de PCA DHFR se utilizó en Escherichia coli para seleccionar dos bibliotecas de secuencias de formación de cremallera de leucina de diseño complementario con 1010 pares potenciales de interacción, de los cuales prácticamente podríamos cubrir 106. Demostramos que la pantalla de PCA seleccionó tanto la especificidad de unión óptima, como la solubilidad y la expresión de cremalleras interactivas (18,25). La característica más importante de este enfoque es que era posible filtrar simultáneamente dos bibliotecas una contra la otra, un proceso que no se logra fácilmente con pantallas híbridas de levadura comparables. La simplicidad de este enfoque y la naturaleza específica de la información obtenida sobre la estrategia de diseño sugieren una amplia utilidad del PCA DHFR en el diseño de proteínas y los experimentos de evolución dirigida. También muestra que el PCA complementa las estrategias de visualización de fagos, ya que toda la selección, optimización y pruebas de rigor se realizan in vivo, lo que hace que este enfoque se ejecute fácilmente.
Aplicación de PCA al Cribado de la Biblioteca de ADNc en Células de mamíferos
Un primer paso para definir la función de un nuevo producto genético es determinar sus interacciones con otros productos genéticos. Sin embargo, un enfoque de cribado puramente basado en la interacción de proteínas (como el híbrido de levadura doble) es limitado, porque solo le dice que dos proteínas interactúan, sin proporcionar ninguna otra información que pueda vincular una proteína a su función. Por lo tanto, hemos demostrado que el PCA se puede utilizar en una estrategia de cribado de la biblioteca de ADNc que combina un cribado simple de interacción de proteínas basado en células con ensayos funcionales específicos que proporcionan una validación inicial de la relevancia biológica de la interacción (9). El primer paso consiste en la detección de interacciones físicas entre el cebo y una biblioteca de proteínas presas codificadas por ADNc, mediante el monitoreo de la reconstitución del reportero de PCA en células vivas intactas. Una característica importante de este primer paso es que las interacciones se pueden detectar directamente y entre proteínas de longitud completa en células en las que la proteína de cebo funciona normalmente, asegurando así que se pueda producir la segmentación subcelular necesaria, las modificaciones postraduccionales y las interacciones con otras proteínas. Obviamente, para la validez experimental, se debe demostrar que los fragmentos de PCA no interfieren con la orientación o modificación de las proteínas. En el segundo paso, la interacción de proteínas se puede validar funcionalmente, de la siguiente manera: en primer lugar, la interacción de proteínas, detectada por el PCA, debe ser perturbada por agentes, como hormonas o inhibidores específicos, que se sabe que modulan la vía bioquímica específica en la que participan las proteínas. Hemos demostrado esto para el PCA DHFR y utilizamos esta propiedad para mapear las vías de señalización en células de mamíferos vivos (16). En segundo lugar, la localización subcelular de la interacción de la proteína, detectada de nuevo por el PCA, podría ser alterada por agentes que modulan la vía. Por lo tanto, la estrategia de cribado basada en PCA combina un paso de cribado simple con ensayos funcionales directos. Nosotros y otros hemos aplicado esta estrategia a la identificación de sustratos o reguladores novedosos de la proteína quinasa serina/treonina, PKB/Akt (9,15,26,27).
Usando PCA como Regla Molecular: Estudios de receptores
Una característica especial de las estrategias de PCA es que, si conocemos la estructura tridimensional de la enzima reportera, es posible predecir con precisión qué tan cerca deben estar los fragmentos para asegurar que la enzima se pliegue correctamente y tenga una actividad medible. Este hecho se puso en práctica para probar un modelo alostérico estructural para la activación del receptor de eritropoyetina dimérica (EPor) utilizando el PCA DHFR, y el enfoque podría extenderse al estudio de transiciones alostéricas en interfaces de proteínas diméricas o multiméricas (19). En el caso del EPor, se demostró que los dominios transmembrana del dímero receptor estaban separados por 73 Å, como se observa en la estructura cristalina del EPor no lagado. Se razonó que si este estado inactivo existía en la membrana de una célula viva, entonces los fragmentos de DHFR fusionados a los terminales C de los dominios transmembrana se plegarían solo si un ligando indujera un cambio de conformación que permitiera que los fragmentos se acercaran lo suficiente para asegurar que se pudiera formar la estructura tridimensional precisa de DHFR (19,28). Esto requeriría que los N terminales de los fragmentos estuvieran separados por 8 Å. La inserción de péptidos enlazadores flexibles entre el dominio transmembrana y los fragmentos de DHFR nos permitió sondear la distancia entre los puntos de inserción del dímero de dominio extracelular y confirmar que se necesitaban enlazadores lo suficientemente largos como para abarcar 73 Å para que el DHFR se plegara de sus fragmentos.
Mapeo de redes bioquímicas
Las maquinarias bioquímicas celulares para metabolismo, cascadas de señalización y ciclo celular son ejemplos de ensamblaje y desmontaje dinámico de complejos macromoleculares. Estos se definen agrupando proteínas que interactúan de acuerdo con sus respuestas similares a un conjunto de perturbaciones (hormonas, metabolitos, inhibidores enzimáticos, etc.).). Las interacciones proteína-proteína se pueden utilizar para vincular una proteína de función desconocida a proteínas que se sabe que participan en un proceso bioquímico conocido. Hemos demostrado que la elaboración de perfiles farmacológicos (control de los efectos de fármacos específicos de la vía y hormonas proteicas en las interacciones proteína-proteína) y la determinación de la ubicación celular de las interacciones proteína-proteína se pueden lograr mediante el uso de PCA (9)(15-17)(26). El análisis de estos resultados permite una representación de cómo evolucionan las redes bioquímicas en el tiempo y el espacio y en respuesta a estímulos específicos. Como prueba de principio, informamos de la aplicación de esta estrategia al mapeo de una vía de transducción de señales mediada por receptores tirosina quinasas (RTK) (16). Los perfiles farmacológicos y la localización celular de las interacciones observadas nos permitieron ubicar cada producto genético en su punto relevante de las vías (Figura 3). A partir de los resultados de nuestro análisis, surgió un mapa de la organización de la red RTK que era coherente con los modelos existentes, pero que también incluía varias interacciones novedosas. La capacidad de monitorear una red de interacciones de proteínas en células vivas que contienen todos los componentes de la vía subyacente estudiada reveló conexiones ocultas, no observadas antes, a pesar del intenso escrutinio de esta red. Los resultados presentados demuestran que la estrategia de PCA tiene las características necesarias para una validación general de la función génica y una estrategia de mapeo de vías. Una aplicación reciente de un conjunto más amplio de acuerdos de asociación y cooperación permitió desarrollar un enfoque general para vincular las acciones de los medicamentos en vías de señalización específicas y detectar actividades imprevistas de los medicamentos (17).
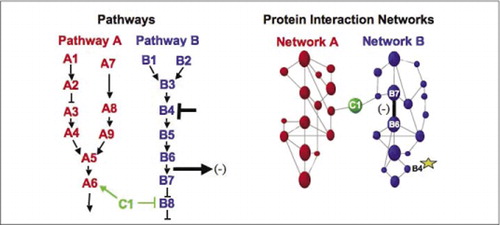
(Izquierda) Las acciones de un agente perturbador inhibitorio que actúa sobre la proteína B4 (barra en T), se detectan aguas abajo por un cambio en la interacción de las proteínas B6 y B7 entre sí (flecha). En este caso, el efecto de la perturbación es una disminución en el número de proteínas que interactúan (-) detectadas por un reportero de esa interacción (señal de salida de la interacción detectada por el centinela de PCA, por ejemplo). Sin embargo, el efecto podría ser igualmente positivo, dependiendo de las consecuencias de la inhibición de la proteína aguas arriba. (Derecha) Dentro de la red de interacción de proteínas para la vía B, una perturbación de la proteína B4 (estrella) de alguna manera se propaga a través de la red para afectar de alguna manera el enlace (barra ancha) entre las proteínas B6 y B7. Esto no implica que la proteína B4 interactúe físicamente con B6 o B7; la propagación de un afecto a través de la red de interacción de proteínas puede deberse a enlaces físicos directos o a procesos enzimáticos no obvios en la red.
Conclusión
El desarrollo y la aplicación del PCA todavía están en curso. Por ejemplo, además de los conjuntos limitados, aunque informativos, de aplicaciones descritos aquí, la estrategia se está aplicando a la detección a gran escala de genomas enteros. Se están explorando problemas más sofisticados de diseño y plegado de proteínas, incluidos estudios de los factores que controlan la selección de secuencias para interacciones óptimas entre proteínas, proteínas y ácidos nucleicos, y proteínas y moléculas orgánicas pequeñas. El PCA es un enfoque experimental muy general y flexible, por lo que deberíamos esperar ver un número creciente de nuevas aplicaciones de esta herramienta básica a la biología molecular y celular en un futuro cercano.
Agradecimientos
Stephen Michnick es titular de la Cátedra de Investigación de Genómica Integrativa de Canadá. La investigación citada de nuestro laboratorio fue financiada por los Institutos Canadienses de Investigación de la Salud.
- 1. Pelletier, J. N., F. X. Campbell-Valois, and S. W. Michnick. 1998. Reensamblaje dirigido por dominio de oligomerización de la dihidrofolato reductasa activa a partir de fragmentos diseñados racionalmente. Proc. Natl. Acad. Sci. USA 95: 12141-12146.Crossref, Medline, CAS, Google Scholar
- 2. Remy, I. y S. W. Michnick. 1999. Selección clonal y cuantificación in vivo de interacciones proteicas con ensayos de complementación de fragmentos proteicos. Proc. Natl. Acad. Sci. USA 96: 5394-5399.Crossref, Medline, CAS, Google Scholar
- 3. Galarneau, A., M. Primeau, L. E. Trudeau, and S. W. Michnick. 2002. Ensayos de complementación de fragmentos de proteínas beta-lactamasas como sensores in vivo e in vitro de interacciones proteicas con proteínas. NAT. Biotechnol. 20:619–622.Crossref, Medline, CAS, Google Scholar
- 4. Wehrman, T., B. Kleaveland, J. H. Her, R. F. Balint, and H. M. Blau. 2002. Interacciones proteína-proteína monitorizadas en células de mamíferos a través de la complementación de fragmentos enzimáticos de beta-lactamasa. Proc. Natl. Acad. Sci. USA 99: 3469-3474.Crossref, Medline, CAS, Google Scholar
- 5. Spotts, J. M., R. E. Dolmetsch, and M. E. Greenberg. 2002. Imágenes de lapso de tiempo de una interacción proteína-proteína dependiente de fosforilación dinámica en células de mamíferos. Proc. Natl. Acad. Sci. USA 99: 15142-15147.Crossref, Medline, CAS, Google Scholar
- 6. Remy, I. y S. W. Michnick. 2006. Ensayo de interacción proteína-proteína altamente sensible basado en Gaussia luciferasa. NAT. Methods 3: 977-979.Crossref, Medline, CAS, Google Scholar
- 7. Ghosh, I., A. D. Hamilton, and L. Regan. 2000. Reensamblaje de proteína dirigida con cremallera de leucina antiparalela: aplicación a la proteína fluorescente verde. J. Am. Chem. Soc. 122:5658–5659.Crossref, CAS, Google Scholar
- 8. Hu, C. D., Y. Chinenov, and T. K. Kerppola. 2002. Visualización de interacciones entre proteínas de la familia bZIP y Rel en células vivas mediante complementación de fluorescencia bimolecular. Mol. Celda 9: 789-798.Crossref, Medline, CAS, Google Scholar
- 9. Remy, I. y S. W. Michnick. 2004. Una estrategia de detección funcional de la biblioteca de ADNc basada en ensayos de complementación de proteínas fluorescentes para identificar nuevos componentes de las vías de señalización. Methods 32: 381-388.Crossref, Medline, CAS, Google Scholar
- 10. Remy, I., F. X. Campbell-Valois, G. Ghaddar, S. Aquin, and S. W. Michnick. 2005. Detection of protein interactions and library screening with protein-fragment complementation assays, p. 637-672. In Protein-Protein Interactions: A Molecular Cloning Manual, 2nd ed. CSH Laboratory Press, Cold Spring Harbor, NY.Google Scholar
- 11. Paulmurugan, R. and S. S. Gambhir. 2003. Monitorización de las interacciones proteína-proteína mediante la complementación asistida por fragmentos de proteína renilla luciferasa sintética dividida. Anal. Chem. 75:1584–1589.Crossref, Medline, CAS, Google Scholar
- 12. Paulmurugan, R., Y. Umezawa, and S. S. Gambhir. 2002. Imágenes no invasivas de las interacciones proteína-proteína en sujetos vivos mediante el uso de estrategias de complementación y reconstitución de proteínas reporter. Proc. Natl. Acad. Sci. USA 99: 15608-15613.Crossref, Medline, CAS, Google Scholar
- 13. Luker, K. E., M. C. Smith, G. D. Luker, S. T. Gammon, H. Piwnica-Worms, and D. Piwnica-Worms. 2004. Cinética de interacciones proteína-proteína reguladas revelada con imágenes de complementación de luciferasa de luciérnaga en células y animales vivos. Proc. Natl. Acad. Sci. USA 101: 12288-12293.Crossref, Medline, CAS, Google Scholar
- 14. Jach, G., M. Pesch, K. Richter, S. Frings, and J. F. Uhrig. 2006. Un mRFP1 mejorado agrega rojo a la complementación de fluorescencia bimolecular. NAT. Methods 3: 597-600.Crossref, Medline, CAS, Google Scholar
- 15. Remy, I., A. Montmarquette, y S. W. Michnick. 2004. PKB / Akt modula la señalización TGF-beta a través de una interacción directa con Smad3. NAT. Cell Biol. 6:358–365.Crossref, Medline, CAS, Google Scholar
- 16. Remy, I. y S. W. Michnick. 2001. Visualización de redes bioquímicas en células vivas. Proc. Natl. Acad. Sci. USA 98: 7678-7683.Crossref, Medline, CAS, Google Scholar
- 17. Macdonald, M. L., J. Lamerdin, S. Owens, B. H. Keon, G. K. Bilter, Z. Shang, Z. Huang, H. Yu, et al.. 2006. Identificación de efectos no deseados y fenotipos ocultos de fármacos en células humanas. NAT. Chem. Biol. 2:329–337.Crossref, Medline, CAS, Google Scholar
- 18. Pelletier, J. N., K. M. Arndt, A. Pluckthun, and S. W. Michnick. 1999. Una selección de biblioteca contra biblioteca in vivo de interacciones optimizadas proteína-proteína. NAT. Biotechnol. 17:683–690.Crossref, Medline, CAS, Google Scholar
- 19. Remy, I., I. A. Wilson, y S. W. Michnick. 1999. Activación del receptor de eritropoyetina por un cambio de conformación inducido por ligandos. Science 283: 990-993.Crossref, Medline, CAS, Google Scholar
- 20. Gegg, C. V., K. E. Bowers, and C. R. Matthews. 1997. Sondeo de unidades plegables independientes mínimas en dihidrofolato reductasa por disección molecular. Proteína Sci. 6:1885–1892.Crossref, Medline, CAS, Google Scholar
- 21. Rossi, F., C. A. Charlton, and H. M. Blau. 1997. Monitorización de las interacciones proteína-proteína en células eucariotas intactas mediante complementación con beta-galactosidasa. Proc. Natl. Acad. Sci. USA 94: 8405-8410.Crossref, Medline, CAS, Google Scholar
- 22. Ozawa, T., S. Nogami, M. Sato, Y. Ohya, and Y. Umezawa. 2000. Indicador fluorescente para detectar interacciones proteína-proteína in vivo basado en el empalme de proteínas. Anal. Chem. 72:5151–5157.Crossref, Medline, CAS, Google Scholar
- 23. Magliery, T. J., C. G. Wilson, W. Pan, D. Mishler, I. Ghosh, A. D. Hamilton, and L. Regan. 2005. Detección de interacciones proteína-proteína con una trampa de reensamblaje de fragmentos de proteína fluorescente verde: alcance y mecanismo. J. Am. Chem. Soc. 127:146–157.Crossref, Medline, CAS, Google Scholar
- 24. Nyfeler, B., S. W. Michnick, and H. P. Hauri. 2005. Captura de interacciones proteicas en la vía secretora de células vivas. Proc. Natl. Acad. Sci. USA 102: 6350-6355.Crossref, Medline, CAS, Google Scholar
- 25. Arndt, K. M., J. N. Pelletier, K. M. Muller, T. Alber, S. W. Michnick, and A. Pluckthun. 2000. Un par de péptidos heterodiméricos en espiral seleccionados in vivo de un conjunto diseñado de biblioteca contra biblioteca. J. Mol. Biol. 295:627–639.Crossref, Medline, CAS, Google Scholar
- 26. Remy, I. y S. W. Michnick. 2004. Regulación de la apoptosis por la proteína Ft1, un nuevo modulador de la proteína quinasa B / Akt. Mol. Celular. Biol. 24:1493–1504.Crossref, Medline, CAS, Google Scholar
- 27. Ding, Z., J. Liang, Y. Lu, Q. Yu, Z. Songyang, S. Y. Lin, and G. B. Mills. 2006. Una pantalla de prueba de complementación de proteínas basada en retrovirus revela socios de unión a AKT1 funcionales. Proc. Natl. Acad. Sci. USA 103:15014–15019.Crossref, Medline, CAS, Google Scholar
- 28. Cody, V., J.R. Luft, E. Ciszak, T.I. Kalman, and J.H. Freisheim. 1992. Crystal structure determination at 2.3 A of recombinant human dihydrofolate reductase ternary complex with NADPH and methotrexate-gamma-tetrazole. Anticancer Drug Des. 7:483–491.Medline, CAS, Google Scholar